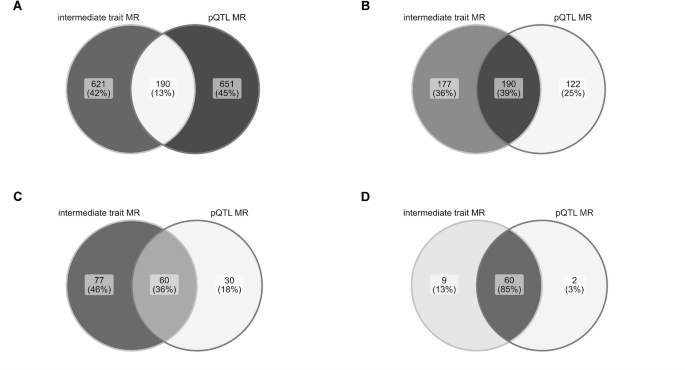
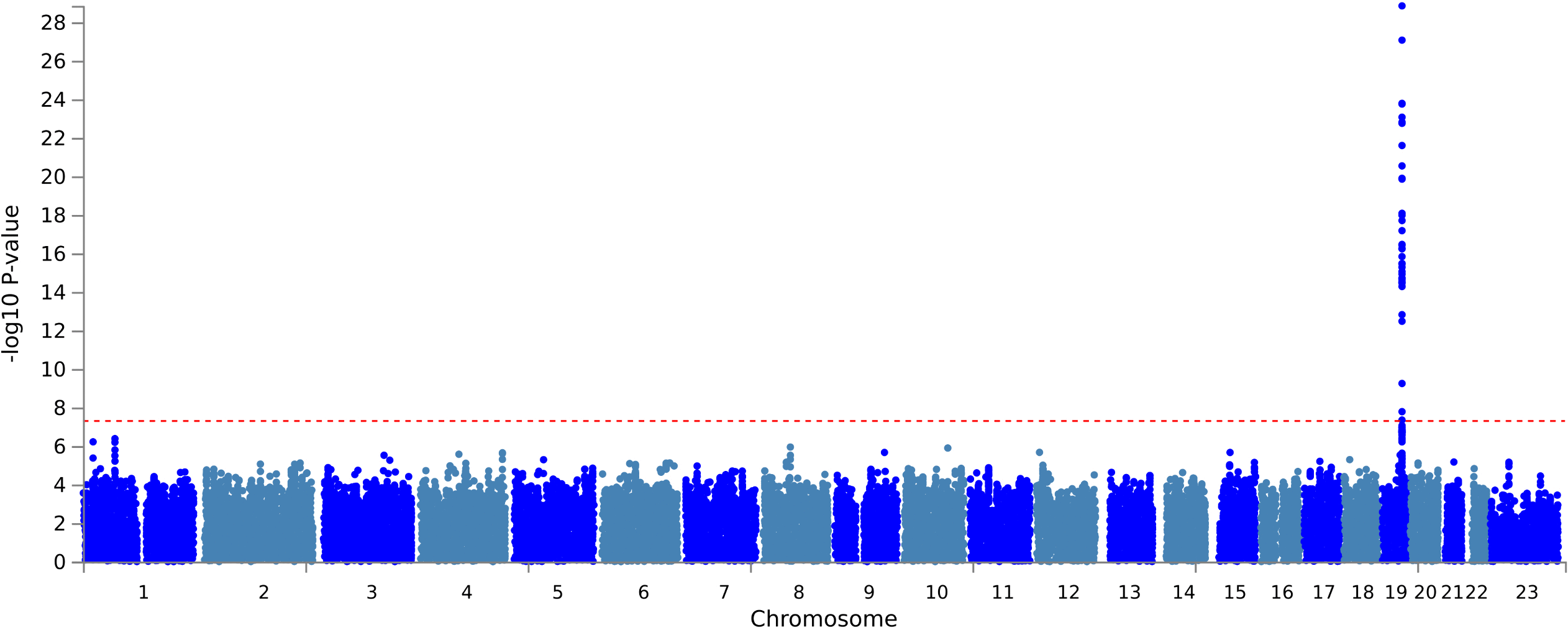
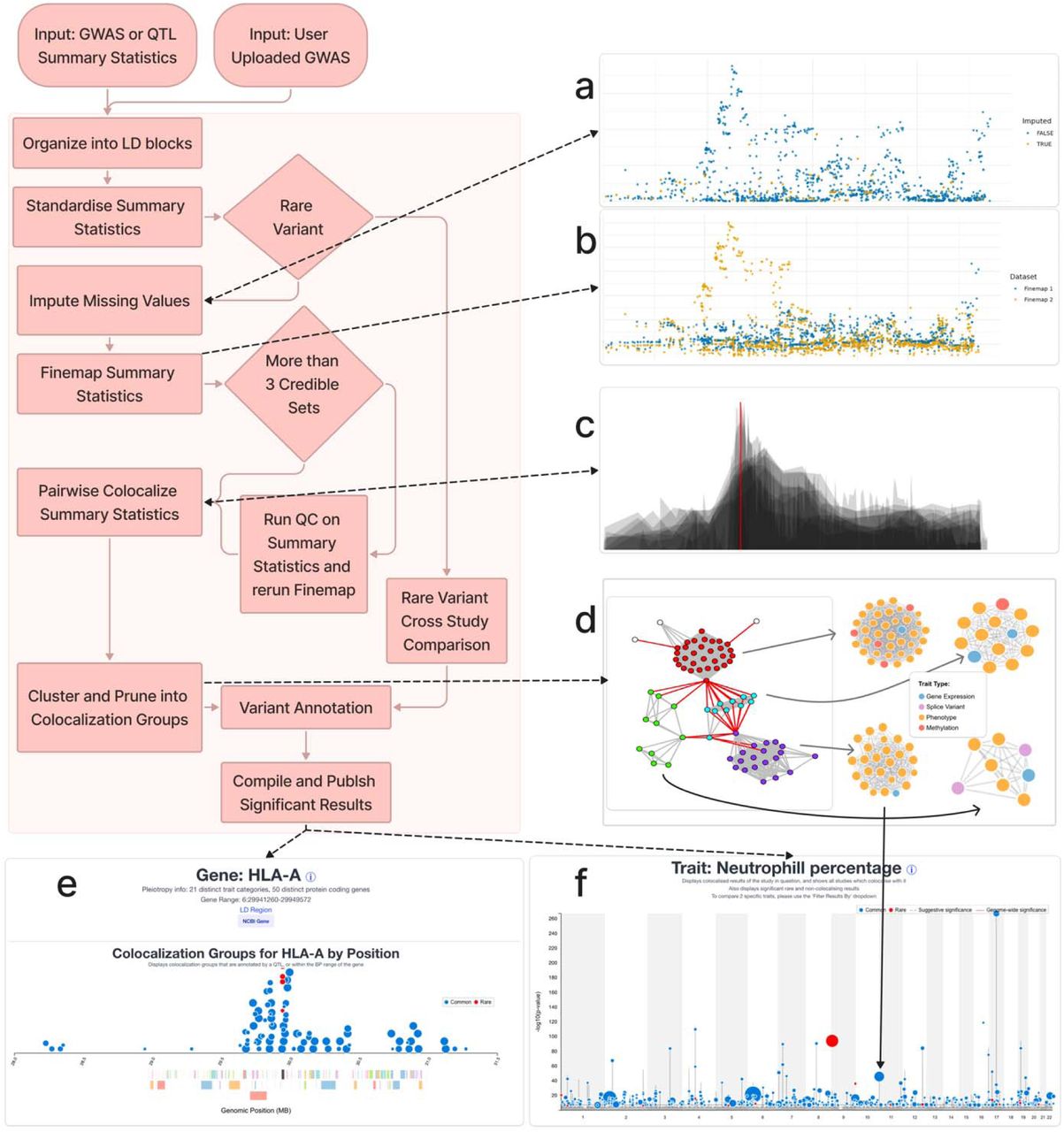
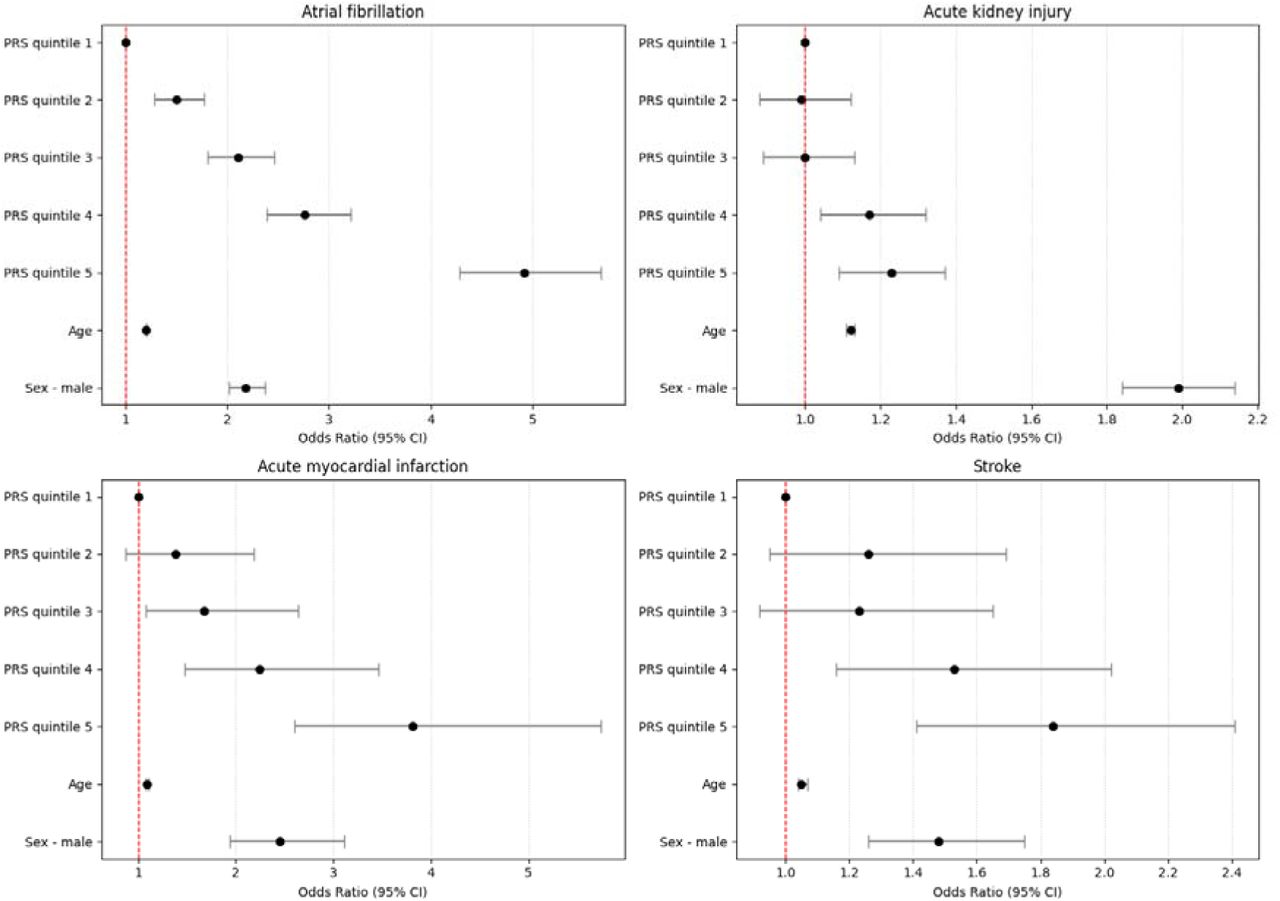
Mapping age-dependent genetic influences on DNA methylation
DNA methylation is often analysed as though genetic effects are stable over time, but development and ageing may change how genetic variation influences the methylome. In a new bioRxiv preprint, Yueying Li and colleagues map age-dependent methylation quantitative trait loci (mQTLs) using repeated DNA methylation measures from birth through adulthood.
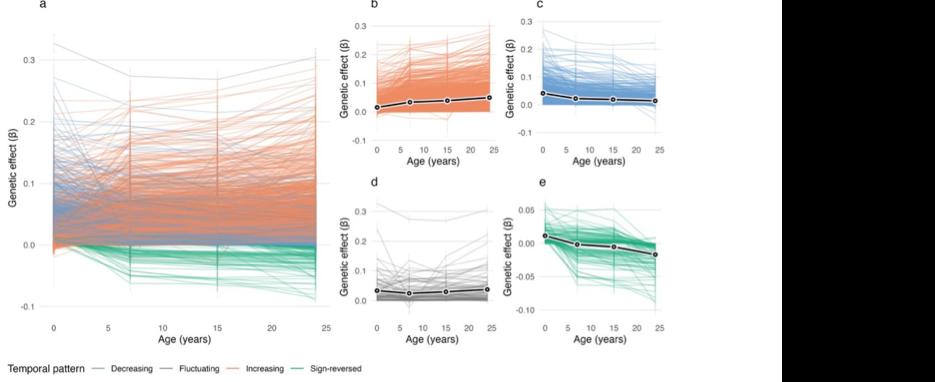
Figure: Trajectories of longitudinal mQTL-CpG associations, grouped by temporal pattern, showing genetic effects that increase, decrease, fluctuate or change sign across age. Source: Li et al., bioRxiv, 2026, Fig. 3 (CC BY 4.0).