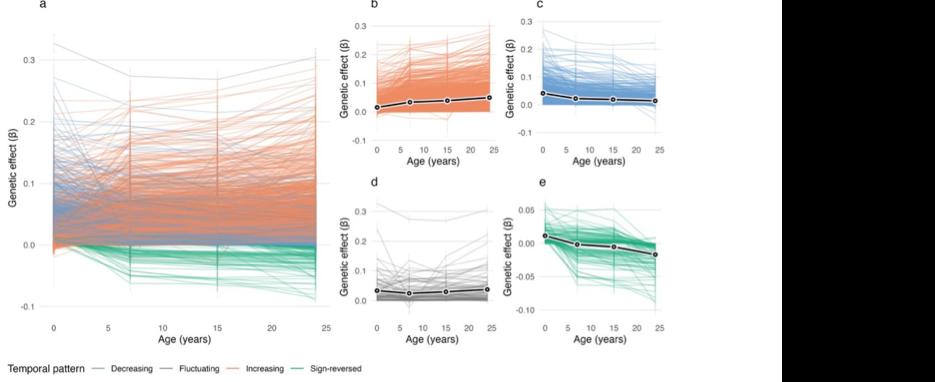
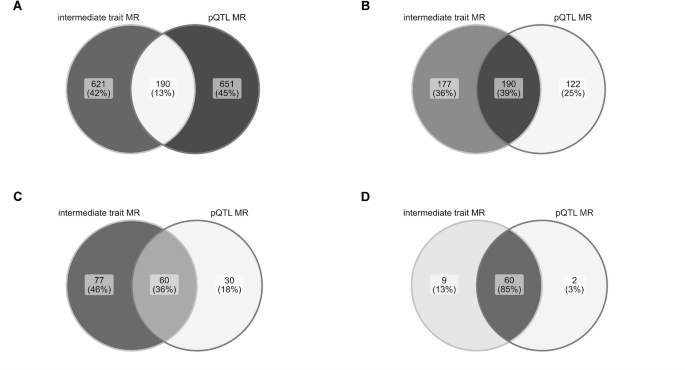
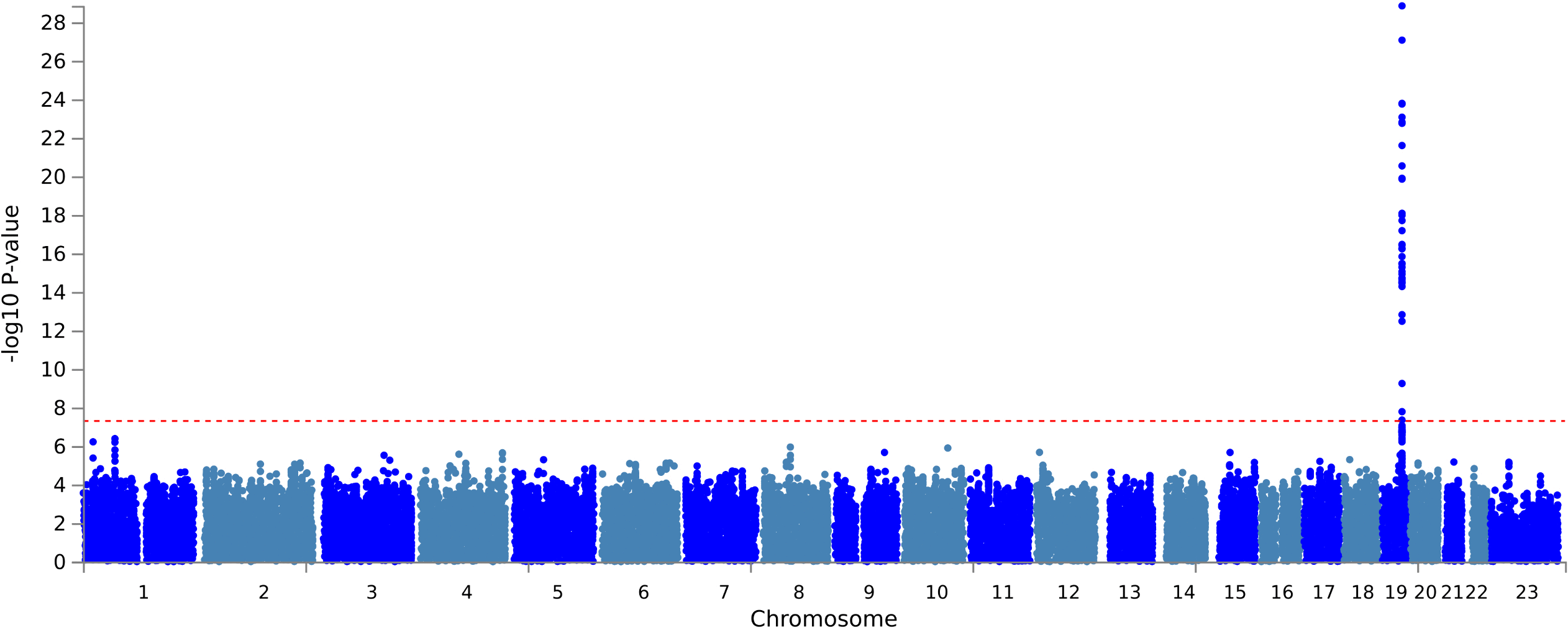
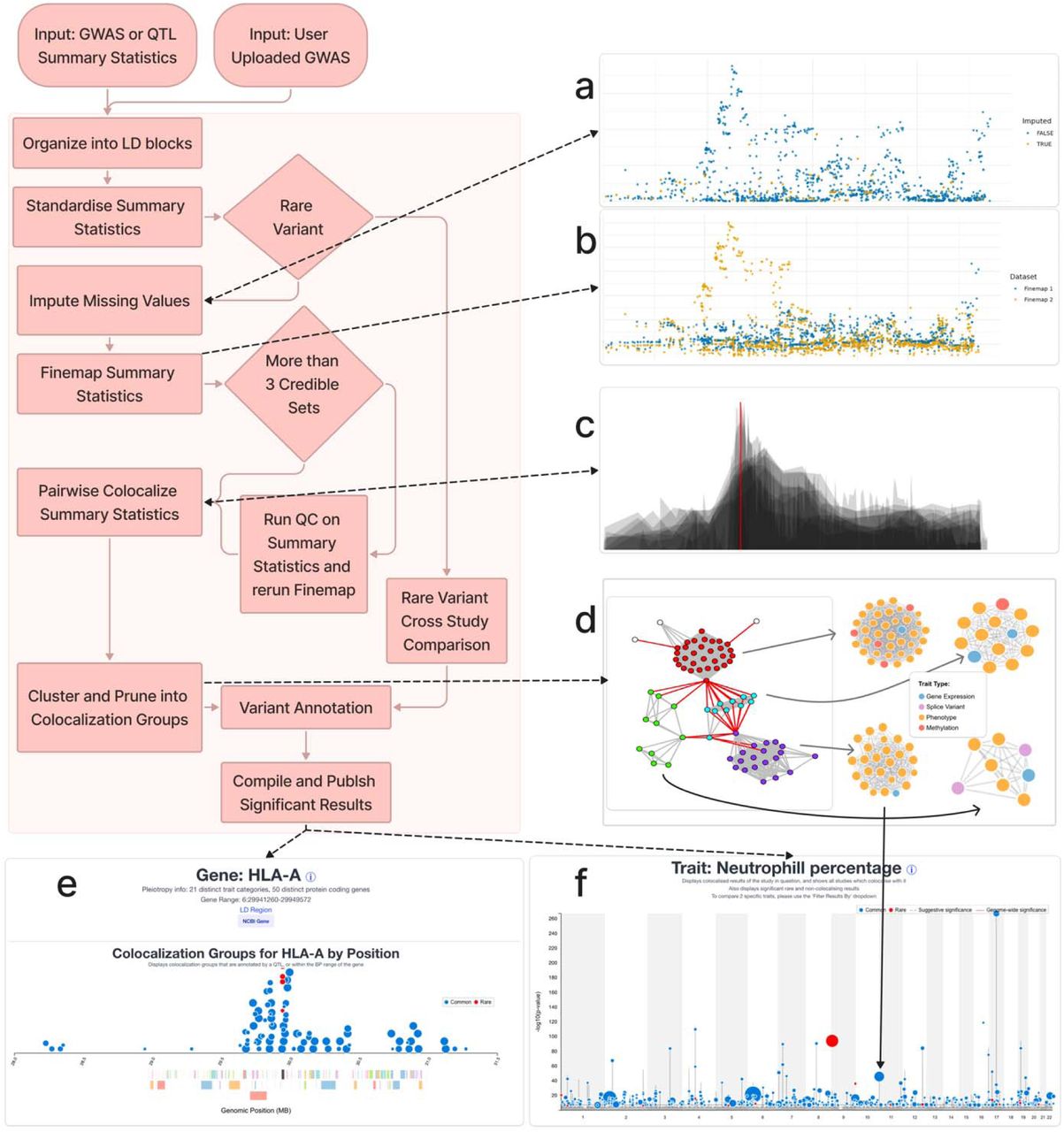
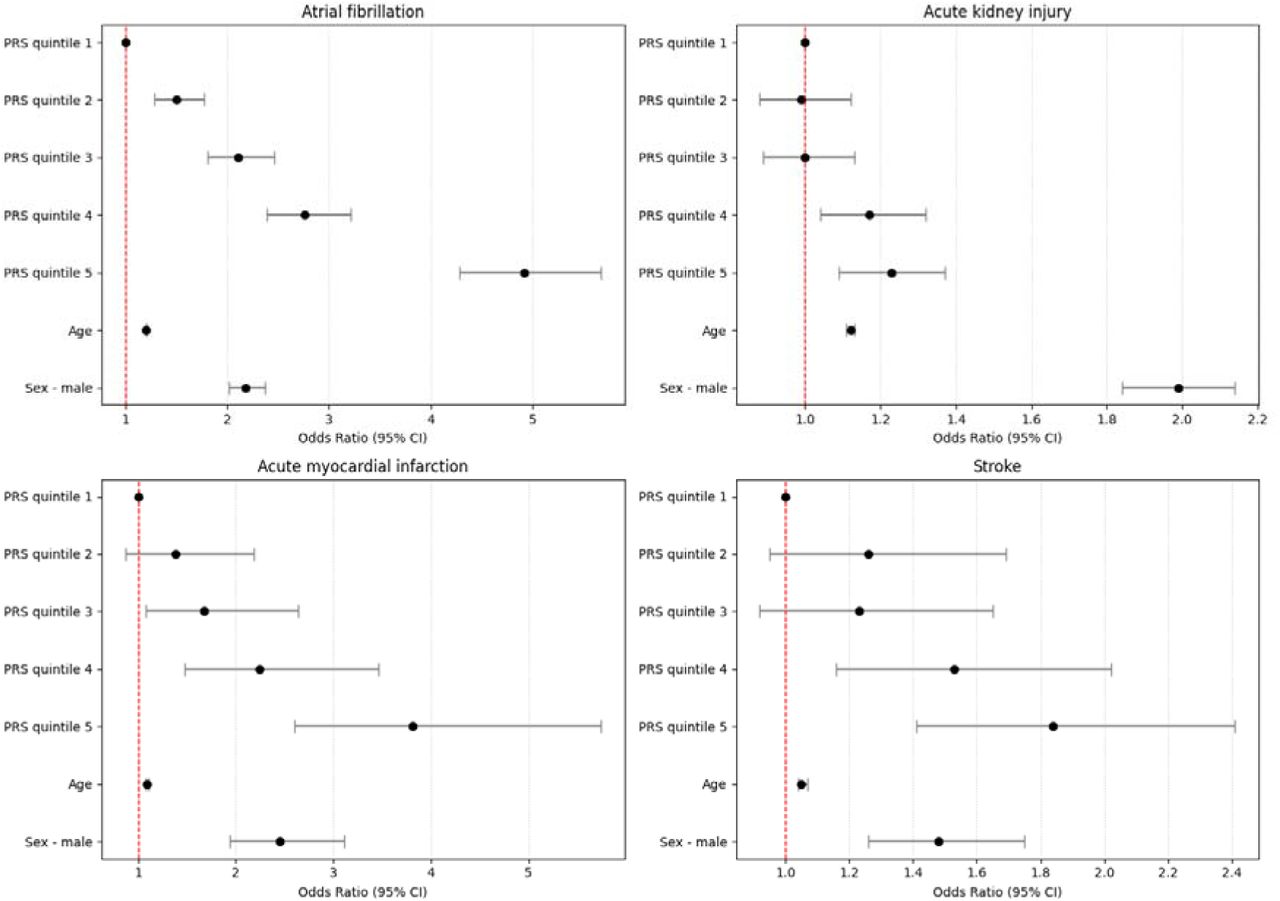
Do historic multiomic profiles improve prediction of postoperative complications?
Biobank metabolomic and proteomic data can predict some long-term disease outcomes, but it is less clear whether historic molecular measurements help predict acute events around surgery. In a new medRxiv preprint, Richard Armstrong and colleagues test whether adding metabolomic and proteomic data improves prediction of postoperative complications after major surgery in UK Biobank.
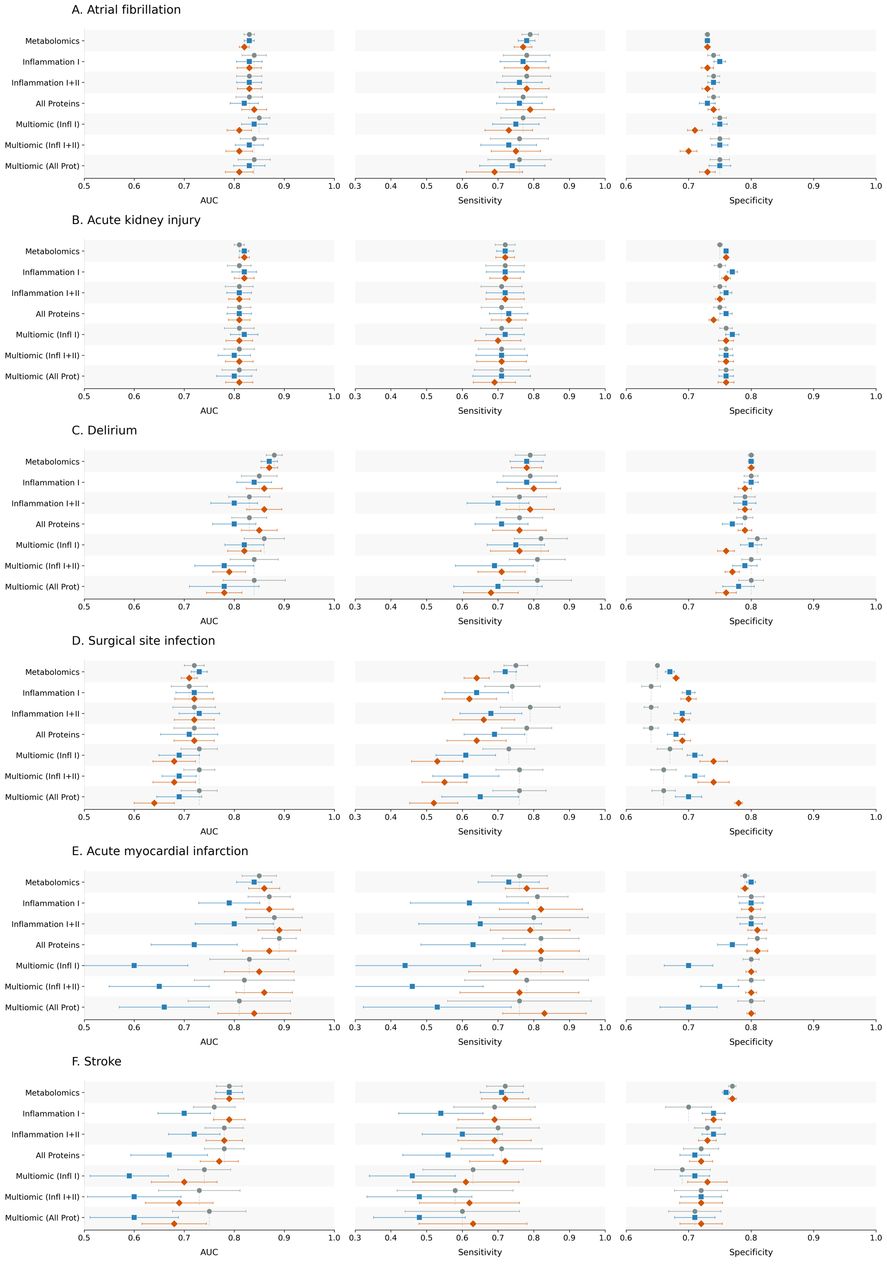
Figure: Model performance across postoperative outcomes and feature sets, comparing clinical baseline models with metabolomic, proteomic and multiomic additions. Source: Armstrong et al., medRxiv, 2026, Fig. 2 (CC BY-NC-ND 4.0).